PAGA¶
[1]:
# load modules
import os
import numpy as np
import pandas as pd
import pickle
# Plotting imports
import matplotlib
import matplotlib.pyplot as plt
import palantir
import scanpy as sc
findfont: Font family ['Raleway'] not found. Falling back to DejaVu Sans.
findfont: Font family ['Lato'] not found. Falling back to DejaVu Sans.
[3]:
%matplotlib inline
Download data¶
Anndata objects with all the data and metadata are publically avaiable at: https://s3.amazonaws.com/dp-lab-data-public/palantir/human_cd34_bm_rep[1-3].h5ad. This notebook use replicate 1 (https://s3.amazonaws.com/dp-lab-data-public/palantir/human_cd34_bm_rep1.h5ad) for illustration.
Description of the anndata object is available at https://s3.amazonaws.com/dp-lab-data-public/palantir/readme
Load data¶
[4]:
# Load the AnnData object
load_ad = sc.read('annadata/human_cd34_bm_rep1.h5ad')
colors = pd.Series(load_ad.uns['cluster_colors'])
ct_colors = pd.Series(load_ad.uns['ct_colors'])
PAGA¶
[5]:
# Start from the counts
ad = sc.AnnData(load_ad.raw.X)
ad.obs_names = load_ad.obs_names
ad.var_names = load_ad.var_names
[6]:
# Preprocess
sc.pp.recipe_zheng17(ad, log=True)
sc.tl.pca(ad)
sc.pp.neighbors(ad, n_neighbors=60)
sc.tl.draw_graph(ad)
sc.tl.louvain(ad, resolution=1.2)
running recipe zheng17
filtered out 27 genes that are detected in less than 1 counts
normalizing by total count per cell
finished (0:00:00.24): normalized adata.X and added
'n_counts_all', counts per cell before normalization (adata.obs)
extracting highly variable genes
the 1000 top genes correspond to a normalized dispersion cutoff of
finished (0:00:00.95)
normalizing by total count per cell
finished (0:00:00.02): normalized adata.X and added
'n_counts', counts per cell before normalization (adata.obs)
... scale_data: as `zero_center=True`, sparse input is densified and may lead to large memory consumption
finished (0:00:00.02)
Note that scikit-learn's randomized PCA might not be exactly reproducible across different computational platforms. For exact reproducibility, choose `svd_solver='arpack'.` This will likely become the Scanpy default in the future.
computing PCA with n_comps = 50
finished (0:00:00.16)
and added
'X_pca', the PCA coordinates (adata.obs)
'PC1', 'PC2', ..., the loadings (adata.var)
'pca_variance', the variance / eigenvalues (adata.uns)
'pca_variance_ratio', the variance ratio (adata.uns)
computing neighbors
using 'X_pca' with n_pcs = 50
computed neighbors (0:00:00.83)
computed connectivities (0:00:06.78)
finished (0:00:00.02) --> added to `.uns['neighbors']`
'distances', distances for each pair of neighbors
'connectivities', weighted adjacency matrix
drawing single-cell graph using layout "fa"
finished (0:00:44.00) --> added
'X_draw_graph_fa', graph_drawing coordinates (adata.obsm)
running Louvain clustering
using the "louvain" package of Traag (2017)
finished (0:00:01.45) --> found 10 clusters and added
'louvain', the cluster labels (adata.obs, categorical)
Graph abstraction¶
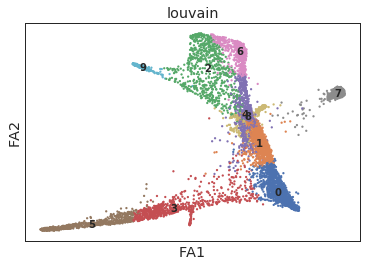
[7]:
ax = sc.pl.draw_graph(ad, color=['louvain'], legend_loc='on data')
[8]:
def hex_to_rgb(value):
value = value.lstrip('#')
lv = len(value)
return tuple(int(value[i:i + lv // 3], 16)/255 for i in range(0, lv, lv // 3))
[9]:
# Palantir colors for comparison
ad.uns['louvain_colors'][0] = hex_to_rgb(colors[0])
ad.uns['louvain_colors'][3] = hex_to_rgb(colors[2])
ad.uns['louvain_colors'][5] = hex_to_rgb(colors[8])
ad.uns['louvain_colors'][7] = hex_to_rgb(colors[5])
ad.uns['louvain_colors'][1] = hex_to_rgb(colors[1])
ad.uns['louvain_colors'][4] = hex_to_rgb(colors[4])
ad.uns['louvain_colors'][6] = hex_to_rgb(colors[3])
ad.uns['louvain_colors'][9] = hex_to_rgb(colors[7])
[10]:
ax = sc.pl.draw_graph(ad, color=['louvain'], legend_loc='on data')
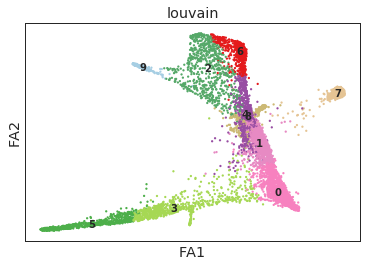
[11]:
sc.tl.paga(ad, groups='louvain')
running PAGA
initialized `.distances` `.connectivities`
finished (0:00:00.46) --> added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns)
Trends¶
[12]:
ad.uns['iroot'] = np.flatnonzero(ad.obs_names == load_ad.obs['palantir_pseudotime'].idxmin())[0]
[13]:
sc.tl.dpt(ad)
WARNING: Trying to run `tl.dpt` without prior call of `tl.diffmap`. Falling back to `tl.diffmap` with default parameters.
computing Diffusion Maps using n_comps=15(=n_dcs)
initialized `.distances` `.connectivities`
computed transitions (0:00:00.04)
eigenvalues of transition matrix
[1. 0.9932567 0.9862846 0.98253083 0.9780972 0.9741326
0.9676509 0.9581074 0.9569015 0.9373291 0.93159527 0.92538345
0.9032845 0.8965749 0.89336187]
finished (0:00:00.25) --> added
'X_diffmap', diffmap coordinates (adata.obsm)
'diffmap_evals', eigenvalues of transition matrix (adata.uns)
initialized `.distances` `.connectivities` `.eigen_values` `.eigen_basis` `.distances_dpt`
computing Diffusion Pseudotime using n_dcs=10
finished (0:00:00.00) --> added
'dpt_pseudotime', the pseudotime (adata.obs)
[14]:
ad_raw = sc.AnnData(load_ad.raw.X)
ad_raw.obs_names = load_ad.obs_names
ad_raw.var_names = load_ad.var_names
# Normalize the data here since raw gets used for trend estimation
sc.pp.log1p(ad_raw)
sc.pp.scale(ad_raw)
ad.raw = ad_raw
... scale_data: as `zero_center=True`, sparse input is densified and may lead to large memory consumption
[15]:
genes = ['CD34', 'MPO', 'IRF8', 'CD79A', 'GATA1', 'ITGA2B'] + \
['CD34', 'SPI1', 'MPO', 'GATA1', 'IRF8', 'CD79B', 'RAG1', 'CEBPG', 'CSF1R']
genes = list(set(genes))
[16]:
trends = pd.Series()
[17]:
paths = [('Ery', [0, 3, 5]), # use the category indices instead of the cluster names
('Mono', [0, 1, 8, 4, 6]),
('CLP', [0, 1, 7]),
('pDC', [0, 1, 8, 4, 2, 9])]
_, axs = plt.subplots(ncols=len(paths), figsize=(6, 2.5), gridspec_kw={'wspace': 0.05, 'left': 0.11})
plt.subplots_adjust(left=0.05, right=0.98, top=0.82, bottom=0.2)
for ipath, (descr, path) in enumerate(paths):
_, data = sc.pl.paga_path(
ad, path, genes,
show_node_names=False,
ax=axs[ipath],
ytick_fontsize=12,
left_margin=0.15,
n_avg=50,
show_yticks=True if ipath==0 else False,
show_colorbar=False,
color_map='Greys',
title='path to\n{} fate'.format(descr[:-1]),
return_data=True,
show=False)
trends[descr] = data
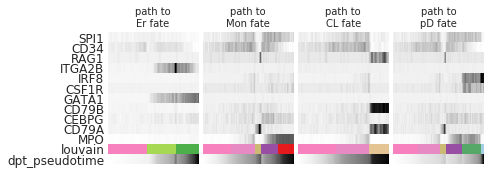
[18]:
genes = ['CD34', 'MPO', 'CD79B', 'GATA1', 'CSF1R', 'ITGA2B']
labels = ['CD34', 'MPO', 'CD79B', 'GATA1', 'CSF1R', 'CD41']
fig = palantir.plot.FigureGrid(6, 6)
layout = ad.obsm['X_draw_graph_fa']
for gene, label, ax in zip(genes, labels, fig):
ax.scatter(layout[:, 0], layout[:, 1], s=3,
c=load_ad[:, gene].X, cmap=matplotlib.cm.Spectral_r)
ax.set_axis_off()
ax.set_title(label)
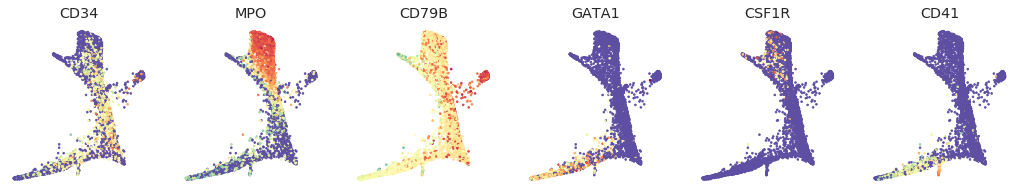
[19]:
from matplotlib.ticker import FormatStrFormatter
[20]:
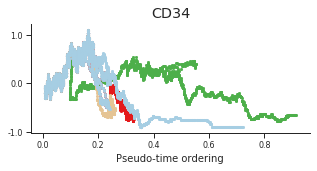
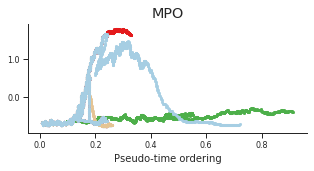
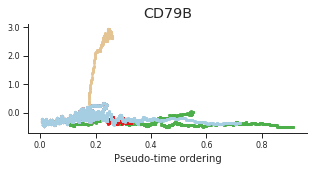
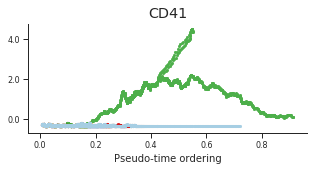
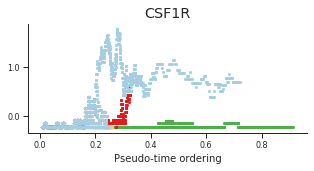
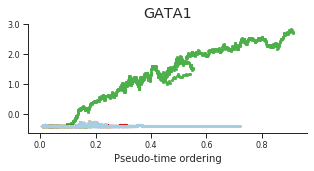
genes = ['CD34', 'MPO', 'CD79B', 'ITGA2B', 'CSF1R', 'GATA1']
labels = ['CD34', 'MPO', 'CD79B', 'CD41', 'CSF1R', 'GATA1']
for gene, label in zip(genes, labels):
fig = plt.figure(figsize=[5, 2])
ax = plt.gca()
for l in trends.keys():
order = trends[l].distance.sort_values().index
bins = np.ravel(trends[l].distance[order])
t = np.ravel(trends[l].loc[order, gene])
# Plot
plt.scatter(bins, t, color=ct_colors[l], s=5)
ax.set_title(label)
ax.set_xlabel('Pseudo-time ordering', fontsize=10)
ax.yaxis.set_major_formatter(FormatStrFormatter('%.1f'))
plt.xticks(fontsize=8)
plt.yticks(fontsize=8)